


COMPUTERIZED SYSTEMS VALIDATION

In this article we are going to discuss about another topic that always raises questions in the pharmaceutical industries’ routine: The Validation of Computerized Systems.
Why to validate?
The main objective of computerized systems validation is to prove through documented tests that the systems properly fulfill their functions in a consistent and safe way.
Where to find orientations?
The main guide for the professionals who validates the systems is the GAMP®5 (Good Automated Manufacturing Practice – A Risk Based Approach to Compliant GxP Computerized Systems), that is currently in the version 5. However, the access to this document é restricted because it is paid and also because it is only available in the English language.
Besides the GAMP, the FDA (Food and Drugs Administration) 21 CFR (Code of Federal Regulation) Part11 standard establishes rules for the use of electronic records in the life sciences industries.
What to validate?
It is very common that, after your first contact with the concept of systems validation, you believe that you need to validate everything that has a software. But, calm down! Before you turn on the emergency siren, let’s understand a bit about the CSV’s scope.
The systems that require validation are called Systems “GxP Relevant” (Good Practices, where the x can be manufacturing, distribution, laboratory, etc.), that is, any and all systems that have an impact on:
• Patient’s health;
• Product quality;
• Data integrity.
It got easier? Probably not that much, right? I understand your anguish, I had also been through it and here is a tip:
Prepare a document to determine the need to validate or not, with the questions below.
If any answer of these questions is “YES”, the system must be validated for having impact on GxP:
• Does the system store data that implies on the products traceability?
• Does the system manage:
• The automated operation of critical production equipment or individual laboratories (compressor, fluid bed, HPLC, dissolvers, etc.)?
• The automated operation of critical utilities generation (purified water, air conditioning, pure air, water for injection, etc.)?
• The registration of presentations, dosages, raw materials, packaging, strengths, batch sizes, production steps, master formulas, etc.?
• The production planning (production orders, batch numbers, raw materials, packaging, etc.)?
• The materials purchase process (providers’ qualification, control of the orders from pre-qualified providers, quantities, strengths, specifications, etc.)?
• The receipt of materials (batch numbers, sampling plan, physical conditions, damage record, etc.)?
• The storage of materials (status, addressing, movements and transfers, etc.)?
• The weighing central (weighing orders, strengths, fractioning, containers, scales, labels and seals, weighing results, operators, products batches, materials batches, etc.)?
• The production control (manufacture orders, process controls, records, operators, materials, batches numbers, used equipment, use and operation sequences, alarms, samples, etc.)?
• The customer service center (complaints, actions, adverse events, etc.)?
• Documentation (emission, distribution, obsolete versions control, training, etc.)?
• Quality systems (out-of-specification results, self-inspection, deviations, changes control, records of results of raw materials analysis, packaging or products, periodic review, etc.)?
• Training program (scope, instructors, attendance lists, certificates, etc.)?
• Equipment (plan and execution of maintenance, plan and execution of calibration, plan and execution of qualification, etc.)?
Anyway, all computerized systems that have a direct or indirect impact with the production of the product or impact on its traceability, need to be validated!!!
Is every system validable?
Great! Now you have already mapped out all the systems that need to be validated, Let’s go to work, right? Not yet! We have one more check to do before elaborating all documentations. The point is: not every system is validable. And now? How to do this mapping? And what to do if the system not fulfill the prerequisites?
Let’s go!
The validation of a computerized system does not resume itself on performing tests to confirm the proper functioning of a software, and its interactions with the hardware. It must also take note to the aspects related to the infrastructure, security, data maintenance, among others.
Firstly, it must follow the compliance requirements with FDA 21 CFR Part11, it means, briefly, the system must include:
• Capacity to store critical data of operations or controls with relevance related to GxP, inviolable electronics files (database with backup and restore);
• Audit Trail – who entered the system, when, what time, what they did, why they did, where they did it?
• Control so that the entries and data modifications are carried out only by authorized persons (security measures, such as the use of passwords, personal code, keys or restricted access to terminals, must be used);
• Different levels of passwords for users and individual users;
• Electronic signature (e.g., login + password to conclude critical steps)
• Strict control of password recovery;
• Ability to record the access attempts by non-authorized persons;
• Ability to record the authorized accesses, including the user, time and date;
• Possibility of printing the electronically stored data.
If any item above is not attended by the system that you are analyzing, it cannot be validated.
And so? Do we just let it go? The answer is NO.
This system must pass through the mitigation process, that basically means to elaborate procedures that cover the failures of the system. For example: the system does not have field to date and sign the electronic records, so for this system it must be used a paper record.
In case the mitigation or upgrade is not possible, the system exchange must be consider, as it cannot be validated.
Valuable tip: if you are acquiring a new system, you must request these requirements to the supplier before the purchase.
How to validate?
Now that your mapping is done, let’s work:
Who is responsible for validating?
The best option is a multidisciplinary team, but every company has your own division. The best option, in my opinion, is the managing by the validation team with the help of the engineering and IT team for the elaboration/revision of the documents, and the tests elaborated and executed by the user or provider.
Computerized Systems Inventory
The inventory of computerized systems is used for identify every existing systems in the company.
The document must contain the following information:
• The system identification, description and version;
• The identification of the system owner;
• The GxP impact assessment (high, medium, low);
• The system status (validated, not validated, etc.);
• The validation report number, if the system has already been validated;
• The interfaces with other systems.
The inventory must contemplate computerized systems, embedded computerized systems, that is, coupled to an equipment (compressor, stove, HPLC, etc.), spreadsheets and systems infrastructure.
Validation Documentation – Life Cycle
We call the “life cycle” the events present throughout the whole time of use of a determined system. These events basically consist of: definition of the specifications of a system for the purchase, acquisition of the system, installation, release, routine use and retirement.
The following figure shows the general approach of the life cycle of computerized systems (V-model).

Source: GAMP®5 (Good Automated Manufacturing Practice – A Risk Based Approach to Compliant GxP Computerized Systems), ISPE, 2008
Both the GAMP5 and the ANVISA Guide detail the exact life cycle required for each type of system and separate as follows:

To better clarify, the following table shows the main documents that must be provided for the validation life cycle of a computerized system.
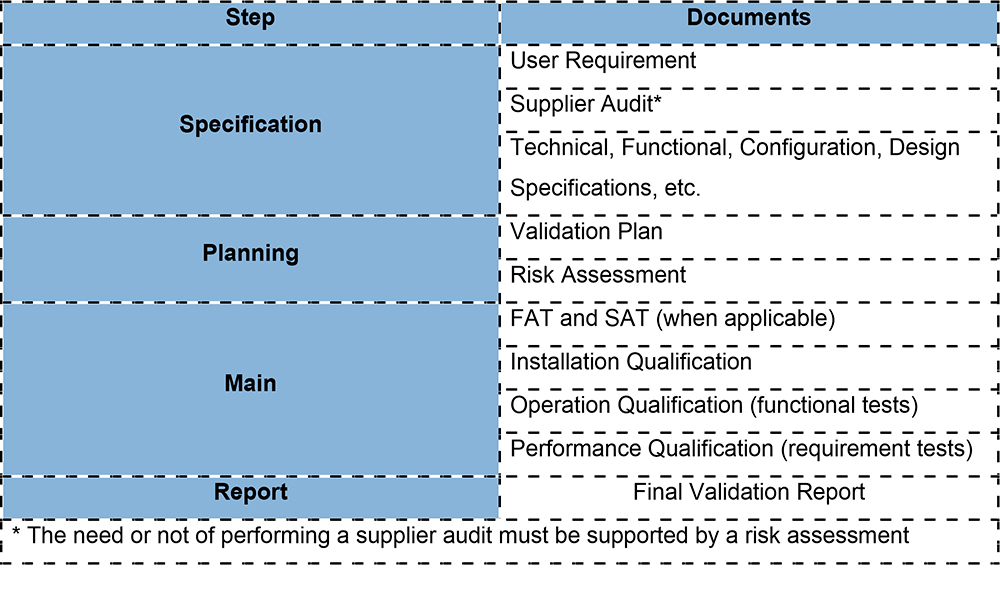
What should be tested?
All the raised risks in the risk assessment must be verified/tested, because, after all, each one has its particularities. However, some risk scenarios are commonly presented and must be tested, examples:
In the Installation Qualification it is needed to verify:
- Procedures for software uses;
- Procedures of backup and restore;
- Anti-virus;
- Procedure of access granting, profile levels and system permissions;
- Hardware that are part of this software (desktops, collectors, printers, etc.)
- Operating and application software and versions;
- Installation in a quality environment to perform the tests.
In the Operation Qualification it is needed to test:
- System behavior in the event of a power outage;
- Permission for created access profiles;
- Access security;
- Quality of electronic records produced by the system including the audit trail;
- Application of electronic signatures;
- System functionality, verify if it operates according to what was described in the functional specification;
- Hardware operation.
In the Performance Qualification it is needed to test:
- Simulate the operation from the beginning to the end of the process and check if it is in accordance to what was requested in the User Requirement.
In some cases (ERP-type systems), the installation and tests can occur in a controlled environment. After the approval of these tests, the system is installed in a production environment, and then the installation must be performed again to ensure that the system that was tested in the tests environment is in production. In addition, after the system release to production, it must be monitored for a period of time stipulated in the Validation Plan to ensure that the system is functioning properly.
If you want to know more about computerized systems, BPx offers training (online and in person), contact us!